Model development
This section walk you through the creation of a mechanistic model from text.
How to use
pasmopy.Text2Model is a useful class to build an ordinary differential equation (ODE) model from a text file describing biochemical systems.
The text file you need to prepare can be divided into three parts:
Note
To write a comment, simply put the hash mark # before your desired comment.
# This is a comment.
Reaction layer
description | parameters | initial conditions
In the reaction layer, you need to describe biochemical reactions.
Each reaction described in the line number i will be converted into ith rate equation.
To specify parameters or initial conditions, you can put those information after |.
If you don’t specify parameters/initial_conditions, they are initialized to 1 and 0, respectively, and the parameter values will be estimated from experimental data.
If you want to set a parameter value to 1 and don’t want to estimate, you can add
constprefix:
# The Hill coefficient is fixed to 1.
TF transcribes mRNA | const n=1
You can impose parameter constraints by specifying line number in the parameter section.
1# Nucleocytoplasmic Shuttling of DUSP
2DUSPc translocates from the cytoplasm to the nucleus <--> DUSPn
3pDUSPc translocates from the cytoplasm to the nucleus <--> pDUSPn |2|
In the example above, you can assume that import and export rates were identical for DUSP (line 2) and pDUSP (line 3).
If the amount of a model species should be held fixed (never consumed) during simulation, you can add
fixedprefix:
# [Ligand] will be held fixed to 10.0 during simulation
Ligand binds Receptor <--> LR | kf = 1e-6, kr = 1e-1 | fixed Ligand = 10.0
To describe more complex rate equations, you can use
@rxnprefix:
@rxn Reactant --> Product: define rate equation here
Please also refer to the following example: cfos_model
Note
The available rules can be found at https://biomass-core.readthedocs.io/en/latest/api/reaction_rules.html.
You can also supply your own terminology in a reaction rule via:
from pasmopy import Text2Model
# Supply "releases" to the reaction rule: "dissociate"
mm_kinetics = Text2Model("michaelis_menten.txt")
mm_kinetics.register_word({"dissociate": ["releases"]})
# Now you can use "releases" in your text, e.g., 'ES releases E and P'
mm_kinetics.convert()
Observable layer (Prefix: @obs)
In the observable layer, you need to specify biomass.observables, which can correlate model simulations and experimental measurements.
You can create an observable by using model parameters (p) and species (u).
For example, if the total amount of SOS bound to EGFR should be the sum of RGS (EGFR-Grb2-SOS) and RShGS (EGFR-Shc-Grb2-SOS) complexes in your model, then you can write as follows:
@obs Total_SOS_bound_to_EGFR: u[RGS] + u[RShGS]
Simulation layer (Prefix: @sim)
In the simulation layer, you can set simulation conditions, e.g, the simulation time span, the initial concentration of model species, etc.
Example:
@sim tspan: [0, 120]
@sim unperturbed: init[EGF] = 0
@sim condition EGF20nM: init[EGF] = 680
@sim condition EGF2nM: init[EGF] = 68
tspan:
Two element vector
[t0, tf]specifying the initial and final times.unperturbed (optional):
Description of the untreated condition to find the steady state.
condition (optional):
Experimental conditions. Use
pandinitto modify model parameters and initial conditions, respectively.
Examples
Michaelis-Menten enzyme kinetics
This example shows you how to build a simple Michaelis-Menten two-step enzyme catalysis model with Pasmopy.
E + S ⇄ ES → E + P
An enzyme, E, binding to a substrate, S, to form a complex, ES, which in turn releases a product, P, regenerating the original enzyme.
Prepare a text file describing biochemical reactions (e.g.,
michaelis_menten.txt)1E + S <--> ES | kf=0.003, kr=0.001 | E=100, S=50 2ES --> E + P | kf=0.002 3 4@obs Substrate: u[S] 5@obs E_free: u[E] 6@obs E_total: u[E] + u[ES] 7@obs Product: u[P] 8@obs Complex: u[ES] 9 10@sim tspan: [0, 100]
Convert the text into an executable model
$ python>>> from pasmopy import Text2Model >>> description = Text2Model("michaelis_menten.txt") >>> description.convert() Model information ----------------- 2 reactions 4 species 3 parameters
Run simulation
>>> from pasmopy import create_model, run_simulation >>> model = create_model("michaelis_menten") >>> run_simulation(model)
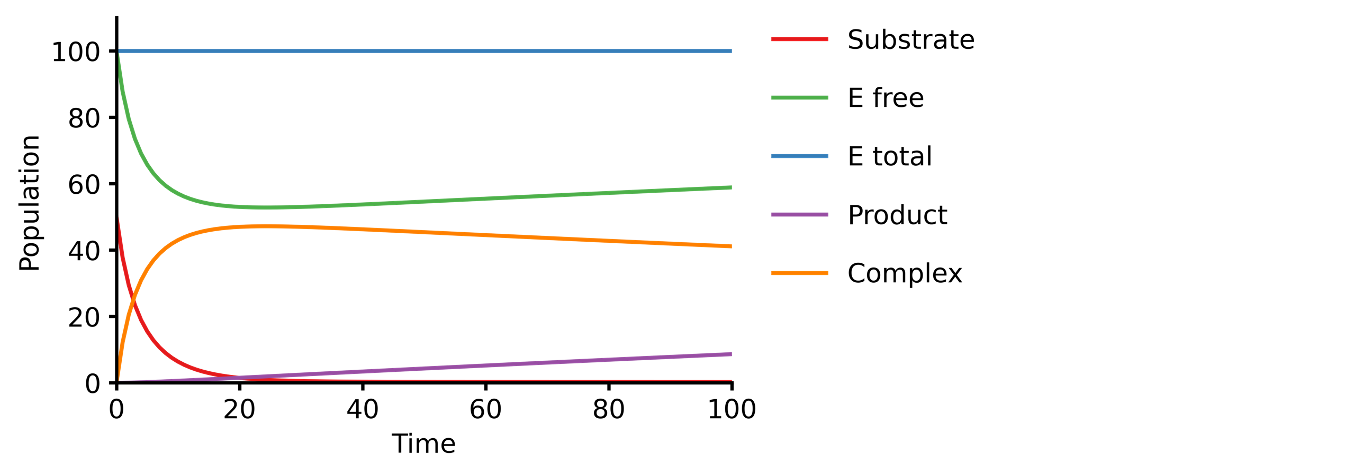
EGF signaling
Below is an example of Pasmopy in action to illustrate EGF signalling pathway.
Reference:
Kholodenko, B. N., Demin, O. V, Moehren, G. & Hoek, J. B. Quantification of short term signaling by the epidermal growth factor receptor. J. Biol. Chem. 274, 30169–30181 (1999). https://doi.org/10.1074/jbc.274.42.30169
Prepare a text describing EGF signaling in hepatocytes (
Kholodenko1999.txt)You can learn how to build the model via
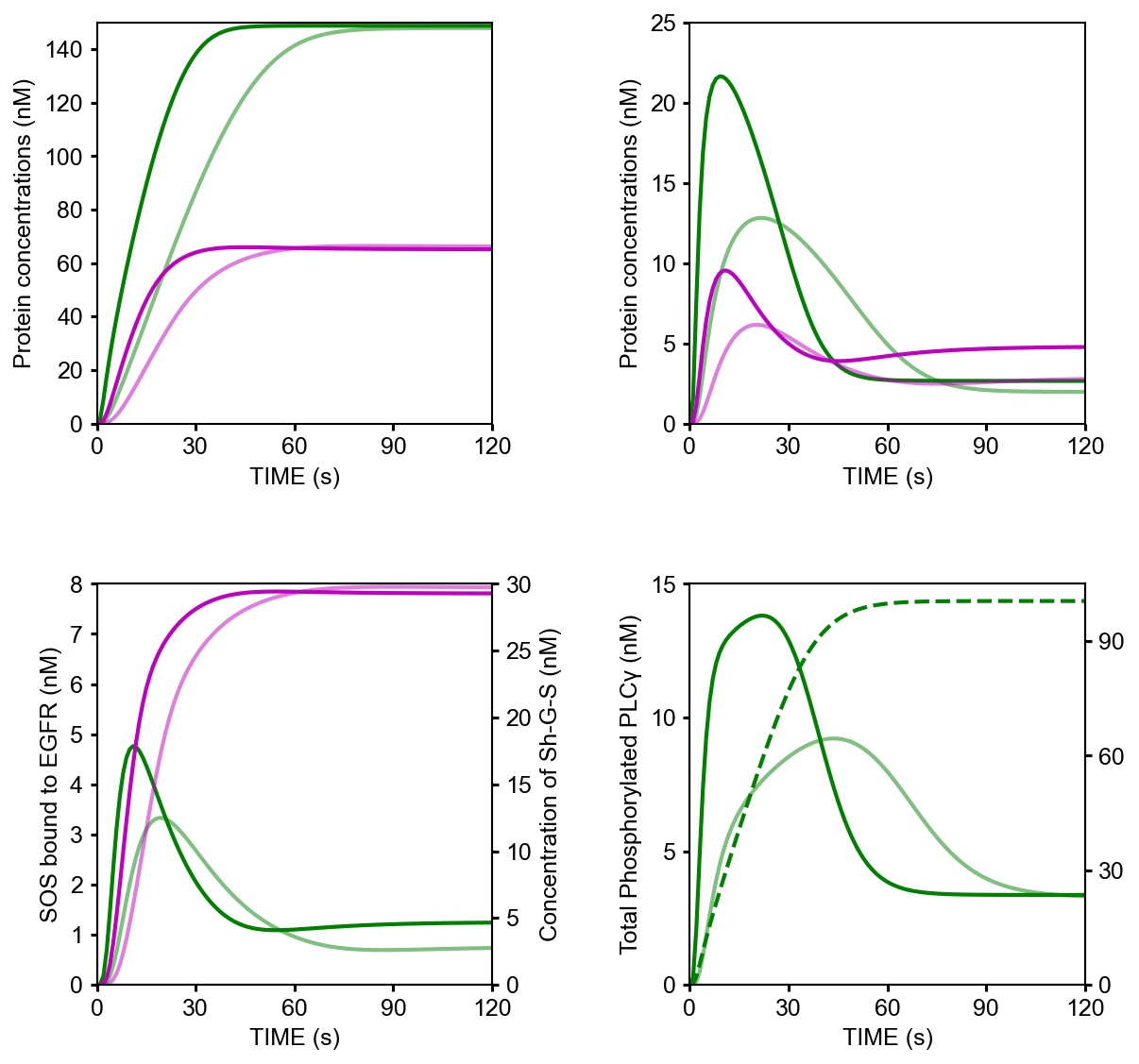
Text2Modelby comparing the desccription below and the network scheme in Figure 1 in the original paper. The description in the line n denotes the n-th reaction in the scheme.1EGF binds EGFR <--> Ra | kf=0.003, kr=0.06 | EGFR=100 2Ra dimerizes <--> R2 | kf=0.01, kr=0.1 3R2 is phosphorylated <--> RP | kf=1, kr=0.01 4RP is dephosphorylated --> R2 | V=450, K=50 5RP binds PLCg <--> RPL | kf=0.06, kr=0.2 | PLCg=105 6RPL is phosphorylated <--> RPLP | kf=1, kr=0.05 7RPLP is dissociated into RP and PLCgP | kf=0.3, kr=0.006 8PLCgP is dephosphorylated --> PLCg | V=1, K=100 9RP binds Grb2 <--> RG | kf=0.003, kr=0.05 | Grb2=85 10RG binds SOS <--> RGS | kf=0.01, kr=0.06 | SOS=34 11RGS is dissociated into RP and GS | kf=0.03, kr=4.5e-3 12GS is dissociated into Grb2 and SOS | kf=1.5e-3, kr=1e-4 13RP binds Shc <--> RSh | kf=0.09, kr=0.6 | Shc=150 14RSh is phosphorylated <--> RShP | kf=6, kr=0.06 15RShP is dissociated into ShP and RP | kf=0.3, kr=9e-4 16ShP is dephosphorylated --> Shc | V=1.7, K=340 17RShP binds Grb2 <--> RShG | kf=0.003, kr=0.1 18RShG is dissociated into RP and ShG | kf=0.3, kr=9e-4 19RShG binds SOS <--> RShGS | kf=0.01, kr=2.14e-2 20RShGS is dissociated into ShGS and RP | kf=0.12, kr=2.4e-4 21ShP binds Grb2 <--> ShG | kf=0.003, kr=0.1 22ShG binds SOS <--> ShGS | kf=0.03, kr=0.064 23ShGS is dissociated into ShP and GS | kf=0.1, kr=0.021 24RShP binds GS <--> RShGS | kf=0.009, kr=4.29e-2 25PLCgP is translocated to cytoskeletal or membrane structures <--> PLCgP_I | kf=1, kr=0.03 26 27# observable layer 28@obs Total_phosphorylated_Shc: u[RShP] + u[RShG] + u[RShGS] + u[ShP] + u[ShG] + u[ShGS] 29@obs Total_Grb2_coprecipitated_with_Shc: u[RShG] + u[ShG] + u[RShGS] + u[ShGS] 30@obs Total_phosphorylated_Shc_bound_to_EGFR: u[RShP] + u[RShG] + u[RShGS] 31@obs Total_Grb2_bound_to_EGFR: u[RG] + u[RGS] + u[RShG] + u[RShGS] 32@obs Total_SOS_bound_to_EGFR: u[RGS] + u[RShGS] 33@obs ShGS_complex: u[ShGS] 34@obs Total_phosphorylated_PLCg: u[RPLP] + u[PLCgP] 35 36# simulation layer 37@sim tspan: [0, 120] 38@sim condition EGF20nM: init[EGF] = 680 39@sim condition EGF2nM: init[EGF] = 68 40@sim condition Absence_PLCgP_transloc: init[EGF] = 680; p[kf25] = 0; p[kr25] = 0
Convert the text into an executable model
$ pythonTo display thermodynamic restrictions, set
show_restrictionstoTrue.>>> from pasmopy import Text2Model >>> description = Text2Model("Kholodenko_JBC_1999.txt") >>> description.convert(show_restrictions=True) Model information ----------------- 25 reactions 23 species 50 parameters Thermodynamic restrictions -------------------------- {9, 12, 10, 11} {15, 18, 21, 17} {18, 22, 20, 19} {17, 24, 12, 19} {23, 24, 20, 15} {23, 12, 22, 21}
The output of Thermodynamic restrictions shows the cyclic pathways in the biochemical reaction network. These detailed balance relations require the product of the equilibrium constants along a cycle to be equal to 1, since at equilibrium the net flux through any cycle vanishes.
Run simulation
>>> from pasmopy import create_model, run_simulation >>> model = create_model("Kholodenko_JBC_1999") >>> run_simulation(model)
Plot simulation results
%matplotlib inline import os import matplotlib.pyplot as plt import numpy as np def plot_simulation_results(res): plt.figure(figsize=(9, 9)) plt.rcParams['font.family'] = 'Arial' plt.rcParams['font.size'] = 12 plt.rcParams['axes.linewidth'] = 1 plt.rcParams['lines.linewidth'] = 2 plt.subplots_adjust(wspace=0.5, hspace=0.4) plt.subplot(2, 2, 1) # ---------------------------------------------------- for obs_name, color in zip( ['Total_phosphorylated_Shc', 'Total_Grb2_coprecipitated_with_Shc'], ['g', 'm'], ): obs_idx = model.observables.index(obs_name) for j, condition in enumerate(['EGF20nM', 'EGF2nM']): plt.plot( model.problem.t, res[obs_idx, j], color=color, alpha=0.5 if condition == 'EGF2nM' else None, ) plt.xlim(0, 120) plt.xticks([30*i for i in range(5)]) plt.ylim(0, 150) plt.xlabel("TIME (s)") plt.ylabel("Protein concentrations (nM)") plt.subplot(2, 2, 2) # ---------------------------------------------------- for obs_name, color in zip( ['Total_phosphorylated_Shc_bound_to_EGFR', 'Total_Grb2_bound_to_EGFR'], ['g', 'm'], ): obs_idx = model.observables.index(obs_name) for j, condition in enumerate(['EGF20nM', 'EGF2nM']): plt.plot( model.problem.t, res[obs_idx, j], color=color, alpha=0.5 if condition == 'EGF2nM' else None, ) plt.xlim(0, 120) plt.xticks([30*i for i in range(5)]) plt.ylim(0, 25) plt.xlabel("TIME (s)") plt.ylabel("Protein concentrations (nM)") ax1=plt.subplot(2, 2, 3) # ------------------------------------------------ ax2 = ax1.twinx() for j, condition in enumerate(['EGF20nM', 'EGF2nM']): ax1.plot( model.problem.t, res[model.observables.index('Total_SOS_bound_to_EGFR'), j], color='g', alpha=0.5 if condition == 'EGF2nM' else None, ) ax2.plot( model.problem.t, res[model.observables.index('ShGS_complex'), j], color='m', alpha=0.5 if condition == 'EGF2nM' else None, ) ax1.set_xlim(0, 120) ax1.set_xticks([30*i for i in range(5)]) ax1.set_xlabel("TIME (s)") ax1.set_ylim(0, 8) ax2.set_ylim(0, 30) ax1.set_ylabel("SOS bound to EGFR (nM)") ax2.set_ylabel("Concentration of Sh-G-S (nM)") ax1=plt.subplot(2, 2, 4) # ------------------------------------------------ ax2 = ax1.twinx() obs_idx = model.observables.index('Total_phosphorylated_PLCg') ax1.plot( model.problem.t, res[obs_idx, model.problem.conditions.index('EGF20nM')], 'g', ) ax1.plot( model.problem.t, res[obs_idx, model.problem.conditions.index('EGF2nM')], 'g', alpha=0.5, ) ax2.plot( model.problem.t, res[obs_idx, model.problem.conditions.index('Absence_PLCgP_transloc')], 'g--', ) ax1.set_xlim(0, 120) ax1.set_xticks([30*i for i in range(5)]) ax1.set_ylim(0, 15) ax1.set_yticks([5*i for i in range(4)]) ax1.set_xlabel("TIME (s)") ax1.set_ylabel("Total Phosphorylated PLCγ (nM)") ax2.set_ylim(0, 105) ax2.set_yticks([30*i for i in range(4)]) plt.show() if __name__ == '__main__': res = np.load(os.path.join(model.path, "simulation_data", "simulations_original.npy")) plot_simulation_results(res)
c-Fos expression dynamics
Please refer to https://biomass-core.readthedocs.io/en/latest/tutorial/cfos.html.